Material de Apoyo de la Clase de la Materia Fisiopatologia
desarrollada el Miercoles 11 de abril de 2012. Uninorte. Asuncion.
Permitida su reproduccion con fines de estudio y guia.

1. GENERALIDADES
El término anemia, utilizado
incorrectamente como un diagnóstico, designa un conjunto de síntomas y signos.
El tipo de anemia define su mecanismo fisiopatológico y su origen, lo que
permite planificar un tratamiento adecuado. Dejar de investigar una anemia leve
es un error grave; su presencia indica una enfermedad subyacente y su gravedad
ofrece poca información sobre su origen o significado clínico verdadero.
Los síntomas y signos de la anemia
representan respuestas cardiovasculares y pulmonares compensadoras según la
gravedad y la duración de la hipoxia tisular. Una anemia grave (p. ej., Hb <7
g/dl) puede asociarse a debilidad, vértigo, cefalea, acúfenos, manchas en el
campo visual, fatiga fácil, mareos, irritabilidad e, incluso, conducta extraña.
Pueden aparecer amenorrea, pérdida de la libido, molestias GI y, en ocasiones,
ictericia y esplenomegalia. Finalmente, puede presentarse insuficiencia cardíaca
y shock.
2. FISIOPATOLOGIA GENERAL DE LAS ANEMIAS
La
anemia es el resultado de una o más combinaciones de tres mecanismos básicos:
pérdida de sangre, eritropoyesis deficiente (producción de hematíes) y
hemólisis excesiva (destrucción de hematíes). La pérdida de sangre debe
ser el primer factor a considerar. Una vez descartado éste, sólo quedan los
otros dos mecanismos. Como la supervivencia de los hematíes es de 120 d, el
mantenimiento de una población estable requiere la renovación diaria de 1/120 de
las células. El cese completo de la eritropoyesis provoca una disminución
aproximada de hematíes del 10%/sem (1%/d). Los defectos de producción tienen
como resultado una reticulocitopenia relativa o absoluta. Cuando las cifras de
hematíes disminuyen a una velocidad >10%/sem (es decir, 500.000
hematíes/ml) sin datos sugestivos de pérdida de sangre, existe una
hemólisis como factor causal.
Un abordaje apropiado en la mayoría de
las anemias debidas a una eritropoyesis deficiente consiste en examinar los
cambios de tamaño y forma de los hematíes. Así, la presencia de anemias
microcíticas sugiere un trastorno
en la síntesis del hem o de la globina (p. ej., deficiencia de hierro [Fe],
talasemia y defectos de la síntesis de Hb relacionados, anemia de las
enfermedades crónicas). Por el contrario, las anemias normocrómicas normocíticas
expresan un mecanismo hipoproliferativo o hipoplásico. Algunas anemias se
caracterizan por macrocitos (hematíes de gran tamaño), lo que indica un defecto
en la síntesis de ADN. Estas anemias se deben generalmente a defectos en el
metabolismo de la vitamina B12 o del ácido fólico o a una
interferencia en la síntesis de ADN por fármacos quimioterápicos
citorreductores. Una respuesta medular adecuada a la anemia se manifiesta por
reticulocitosis o policromatofilia en sangre periférica.
De forma semejante, algunos mecanismos
comunes de aumento de la destrucción (p. ej., secuestro esplénico, hemólisis
mediada por anticuerpos, función defectuosa de la membrana eritrocitaria o Hb
anómala) ayudan en gran medida al diagnóstico diferencial de las anemias
hemolíticas.
3. CLASIFICACION GENERAL DE LAS ANEMIAS

((hacer click para agrandar)
a. ANEMIAS MACROCITICAS
ANEMIA MACROCITICA MEGALOBLASTICA
La forma no megaloblástica de la anemia
macrocítica (es decir, VCM >95 fl/célula) es heterogénea, de manera que los
cambios macrocíticos periféricos no se asocian a las características de
laboratorio, bioquímicas y clínicas típicas de la megaloblastosis.
La anemia macrocítica no megaloblástica
aparece en diversos estados clínicos, aunque no todos ellos se comprenden. La
macrocitosis con exceso de membrana eritrocitaria se observa en pacientes con
hepatopatía crónica que presentan una esterificación defectuosa del colesterol.
Dado que el moldeado de la membrana eritrocitaria se realiza en el bazo tras
liberarse las células desde la médula ósea, los hematíes pueden ser ligeramente
macrocíticos después de una esplenectomía, si bien estas alteraciones no se
asocian a anemia. El consumo crónico de alcohol también se ha relacionado con
índices eritrocitarios macrocíticos (generalmente VCM entre 95 y 105 fl/célula);
estos cambios no se deben a deficiencia de ácido fólico ni a otros mecanismos
metabólicos reconocibles. En la anemia aplásica también se observa una
macrocitosis moderada (v. más arriba), sobre todo cuando se produce la
recuperación. En cada una de estas circunstancias, la anemia se relaciona con
mecanismos diferentes a los de la macrocitosis y la médula no es megaloblástica.
Una clave añadida para establecer el origen de la macrocitosis consiste en la
ausencia de macroovalocitos típicos en la extensión periférica y de aumento de
la ADE, datos típicos de la anemia megaloblástica clásica.
Finalmente, los cambios macrocíticos son
habituales en la mielodisplasia, en que la heterogeneidad celular se pone de
manifiesto por una ADE elevada y una marcada anisocitosis. La médula ósea
contiene precursores eritrocitarios megaloblastoides (también frecuentes en las
hepatopatías avanzadas), que expresan unos patrones densos y groseros de
cromatina nuclear y que son distintos de las alteraciones observadas en la
anemia megaloblástica típica.
Los estados megaloblásticos se deben a la
síntesis defectuosa de ADN. La síntesis de ARN continúa y, como resultado, se
produce un aumento de la masa y la maduración citoplasmáticas. La circulación
recibe hematíes macroovalocíticos y todas las células presentan dispoyesis,
caracterizada porque la maduración citoplasmática es mayor que la nuclear,
produciéndose el megaloblasto en la médula. La dispoyesis incrementa la
destrucción intramedular de las células (eritropoyesis ineficaz), originándose
hiperbilirrubinemia indirecta e hiperuricemia. Como están afectadas todas las
líneas celulares, además de la anemia, pueden aparecer leucopenia y
trombocitopenia, aunque suelen tardar en desarrollarse. Otro hallazgo
característico del estado megaloblástico lo constituye la reticulocitopenia
debida a la producción defectuosa de hematíes. La hipersegmentación de los
neutrófilos polimorfonucleares también es un hallazgo típico, cuyo mecanismo de
producción se desconoce. Además del reconocimiento morfológico de los cambios
megaloblásticos, puede utilizarse la prueba de supresión de desoxiuridina para
demostrar la síntesis defectuosa de ADN a nivel bioquímico.
Los mecanismos más
frecuentes que causan estados megaloblásticos incluyen la utilización
deficitaria o defectuosa de vitamina B12 o ácido fólico, los fármacos
citotóxicos (generalmente antineoplásicos o inmunodepresores) que alteran la
síntesis de ADN y una forma neoplásica autónoma rara, el síndrome de Di
Guglielmo, que se considera una mielodisplasia en transformación a leucemia
mieloide aguda. La identificación de la etiología y de los mecanismos
fisiopatológicos de las anemias megaloblásticas resulta crucial.
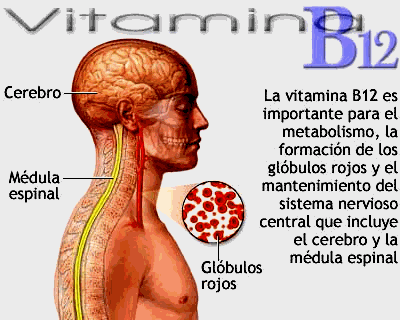
ANEMIA POR DEFICIT DE VITAMINA B12. ANEMIA PERNICIOSA
La molécula de vitamina
B12 consta del nucleótido 5,6-dimetilbencimidazol unido en ángulo
recto con un anillo tetrapirrólico con un átomo de cobalto (el núcleo
corrínico). En la naturaleza existen diversas cobalaminas (componentes de
la vitamina B12) que sólo varían en el radical unido al átomo de
cobalto.
La metilcobalamina
(MeCbl) y la adenosilcobalamina (AdoCbl), dos coenzimas de cobalamina
fisiológicas, realizan las funciones bioquímicas de la B12. La MeCbl
actúa en el metabolismo del ácido nucleico y es el cofactor que interviene en la
síntesis de ADN defectuoso. La AdoCbl sirve como un sistema de recogida para el
catabolismo de los aminoácidos alifáticos, las membranas lipídicas y los
precursores de propionato y es posible que sea el cofactor implicado en la
síntesis y reparación de la mielina alterada.
La vitamina B12
está presente en la carne y los alimentos con proteínas animales. Su absorción
es compleja; se lleva a cabo en el íleon terminal y requiere la presencia de
factor intrínseco, una secreción de las células parietales de la mucosa
gástrica, para transportar la vitamina a través de la mucosa intestinal. La
vitamina B12 alimentaria se une a proteínas fijadoras (fijadoras R)
de la saliva que protegen a la B12 en el medio ácido gástrico. Cuando
este complejo B12 (B12-fijadoras R) se introduce en el
intestino delgado, unas enzimas pancreáticas lo escinden y la vitamina
B12 se une al factor intrínseco.
La vitamina B12 está presente
en el plasma unida a proteínas
específicas, las transcobalaminas I y II. La transcobalamina I es una forma de
depósito, en tanto que la transcobalamina II es la proteína transportadora de
B12 fisiológica. La concentración plasmática normal de vitamina
B12 oscila entre 200 y 750 pg/ml (150-550 pmol/l), lo que sólo
representa alrededor del 0,1% del contenido total del organismo, la mayoría del
cual se localiza en el hígado. La excreción es principalmente biliar y, en menor
grado, renal. La pérdida diaria total es de 2-5 mg; se produce cierta
reutilización enterohepática.
Debido a la lenta tasa de utilización y a
los considerables depósitos de vitamina B12, su deficiencia
(depósitos tisulares <0,1 mg y valor sérico <150 pg/ml [110 pmol/l]) tarda
en aparecer entre varios meses y años. Los depósitos hepáticos de B12
suelen bastar para satisfacer las necesidades fisiológicas durante 3-5 años en
ausencia de factor intrínseco y de meses a un año en ausencia de capacidad de
reabsorción enterohepática. No obstante, cuando los depósitos hepáticos son
limitados y la demanda por el crecimiento es elevada, las alteraciones
hematológicas y neurológicas pueden aparecer con mayor rapidez (p. ej., niños
lactantes de madres vegetarianas).
La disminución de la
absorción de vitamina B12 es el principal mecanismo fisiopatológico y
puede deberse a varios factores. La anemia causada por
deficiencia de vitamina B12 también suele denominarse anemia
perniciosa. Clásicamente, el término anemia perniciosa expresa la
deficiencia de B12 producida por pérdida de la secreción de factor
intrínseco. La
competencia por la vitamina B12 disponible y la escisión del factor
intrínseco pueden ocurrir en el síndrome del asa ciega (debido al empleo
bacteriano de B12) o en las infestaciones por cestodos. Las áreas de
absorción ileal pueden faltar de forma congénita o destruirse por enteritis
regional inflamatoria o resección quirúrgica. Causas menos frecuentes de
disminución de la absorción de B12 incluyen la pancreatitis crónica,
los síndromes de malabsorción, la administración de ciertos fármacos (p. ej.,
quelantes orales del calcio, ácido aminosalicílico, biguanidas), la ingestión
inadecuada de B12 (generalmente en vegetarianos) y, en muy raras
ocasiones, el aumento del metabolismo de la B12 en el hipertiroidismo
de larga duración. Una causa muy habitual de deficiencia de B12 en la
población anciana es la absorción inadecuada de B12 unida a alimentos
en ausencia de cualquiera de los mecanismos anteriores; la vitamina
B12 pura se absorbe, pero la liberación y la absorción de la
B12 unida a alimentos son defectuosas.
La anemia generalmente se desarrolla de
manera insidiosa y progresiva a medida que se agotan los depósitos hepáticos de
B12. A menudo, es más intensa de lo que cabría esperar por los
síntomas, porque su lenta evolución permite una adaptación fisiológica. En
ocasiones se palpan esplenomegalia y hepatomegalia. Pueden estar presentes
diversas manifestaciones GI, como anorexia, estreñimiento y diarrea
intermitentes y dolor abdominal mal localizado. La glositis, descrita
generalmente como una quemazón sobre la lengua, puede ser un síntoma temprano.
Es frecuente una pérdida de peso considerable.
Puede haber afectación neurológica
incluso en ausencia de anemia. Este hecho se comprueba sobre todo en pacientes
mayores de 60 años. Los nervios periféricos son los que se afectan con mayor
frecuencia, seguidos de la médula espinal. Los síntomas neurológicos preceden
algunas veces a las alteraciones hematológicas (e incluso ocurren en su
ausencia, en especial si se ha administrado ácido fólico).
En las fases iniciales se detecta una
pérdida periférica de la sensibilidad posicional y vibratoria en las
extremidades, junto con debilidad leve o moderada y pérdida de reflejos. En
fases posteriores aparecen espasticidad, signo de Babinski, mayor pérdida de la
sensibilidad propioceptiva y vibratoria en las extremidades inferiores y ataxia.
La sensibilidad táctil, algésica y térmica se alteran con menos frecuencia. Las
extremidades superiores se afectan más tarde y con menos regularidad que las
inferiores. Algunos pacientes también muestran irritabilidad y depresión
moderada. Puede desarrollarse ceguera para los colores azul y amarillo. En los
casos avanzados puede surgir paranoia (demencia megaloblástica), delirio,
confusión, ataxia espástica y, en ocasiones, hipotensión postural.
ANEMIA POR DEFICIT DE ACIDO FOLICO
Numerosos tejidos vegetales y animales
contienen ácido fólico. En la forma tetrahidrato, los
folatos actúan como coenzimas en procesos en los que existe transferencia de una
unidad de carbono (p. ej., en la biosíntesis de nucleótidos purínicos y
pirimidínicos), en conversiones de aminoácidos (p. ej., de histidina a ácido
glutámico a través del ácido formiminoglutámico) y en la síntesis y utilización
de formatos.
La absorción se lleva a cabo en el
duodeno y el yeyuno proximal. En las células epiteliales, los poliglutamatos de
los alimentos se reducen hasta dihidrofolatos y tetrahidrofolatos. Se unen a
proteínas y se transportan como metiltetrahidrofolato. Los valores séricos
oscilan entre 4 y 21 ng/ml (9-48 nmol/l) y son un fiel reflejo de la ingestión
dietética. El folato eritrocitario (valores normales, 225-640 ng/ml de sangre
total [510-1.450 nmol/l], corregido a un Hto del 45%) constituye un indicador
más adecuado del estado tisular de folato. El folato total del organismo se
aproxima a 70 mg, localizándose la tercera parte en el hígado. Alrededor del 20%
del folato ingerido se excreta sin haberse absorbido, junto con 60-90
mg/d
no reabsorbidos por la bilis.
La cocción prolongada destruye los
folatos, que son abundantes en ciertos alimentos como vegetales de hoja verde,
levaduras, hígado y setas. En ausencia de ingestión, los depósitos hepáticos
sólo proporcionan suministro durante 2-4 meses. Es habitual la ingestión
dietética limitada de ácido fólico. El alcohol interfiere en su metabolismo
intermediario, absorción intestinal y circulación enterohepática. Por esta
razón, las personas que siguen una dieta carencial (p. ej., "té y tostadas",
alcohólicos crónicos) son propensas a desarrollar una anemia macrocítica por
déficit de folato, al igual que aquellos que padecen una hepatopatía crónica.
Dado que el feto obtiene el ácido fólico por suministro materno, las mujeres
gestantes son susceptibles de desarrollar una anemia megaloblástica.
La malabsorción intestinal es otra causa
frecuente de deficiencia de folato. El déficit de folato puede desarrollarse en pacientes
tratados con anticonvulsivantes o anticonceptivos orales durante períodos
prolongados debido a la disminución de la absorción, así como en individuos en
tratamiento con antimetabolitos (metotrexato) y fármacos antimicrobianos (p.
ej., trimetoprim/sulfametoxazol) que alteran el metabolismo del folato.
Finalmente, el aumento de la demanda de folato se produce en la gestación y la
lactancia, en las anemias hemolíticas crónicas (sobre todo congénitas), en la
psoriasis y en la diálisis crónica.
Las manifestaciones clínicas principales
son las propias de la anemia. La deficiencia de folato es indistinguible de la
de vitamina B12 por los hallazgos en sangre periférica y en médula
ósea, pero no se observan las lesiones neurológicas propias del déficit de
B12. El folato es fundamental en la formación del sistema nervioso
durante los períodos fetal y neonatal. Cuando no se ingiere folato durante el
embarazo pueden surgir defectos del tubo neural con alteraciones neurológicas
graves. Otros síntomas neurológicos poco frecuentes (síndrome de piernas
inquietas del embarazo) también se han relacionado con la deficiencia de folato.
ANEMIA POR DEFICIT DE VITAMINA C
La deficiencia de vitamina C (ácido
ascórbico) suele asociarse a
anemia hipocrómica, pero ésta puede ser normocítica o microcítica (con pérdida
crónica de sangre). En ocasiones, la anemia macrocítica por deficiencia de
vitamina C se asocia a déficit de ácido fólico y su corrección requiere vitamina
C (500 mg/d) y ácido fólico (dosis mencionadas anteriormente).
b. ANEMIAS MICROCITICAS O HIPOCROMICAS
Son las anemias en las que los glóbulos rojos tienen disminuido, por el contrario su contenido en hemoglobina mas que disminuido su número. El HCM viene a ser inferior al normal, estando los glóbulos rojos decolorados. De este tipo de anemias son:
- La anemia microcítica o ferropénica.
- Talasemia.
- Algunas anemias sideroblásticas.
- Intoxicación por plomo (algunas veces).
- Intoxicación por aluminio (rara vez).
- Las anemias producidas por enfermedades crónicas (a veces).
La síntesis deficiente o defectuosa del
hem o de la globina produce una población de hematíes microcíticos. No obstante,
los cambios iniciales pueden ser mínimos.
El Fe se distribuye en los
compartimientos metabólico activo y de depósito. El Fe total del organismo es
aproximadamente de 3,5 g en los varones adultos sanos y de 2,5 g en las mujeres;
la diferencia guarda relación con el tamaño corporal y la ausencia habitual de
un depósito significativo de Fe en las mujeres. El contenido aproximado en el
depósito activo de un individuo medio es de 2.100 mg en Hb, 200 mg en
mioglobina, 150 mg en enzimas (hem y no hem) tisulares y 3 mg en el
compartimiento de transporte de Fe. El Fe se almacena en las células de los
tejidos como ferritina (700 mg) y hemosiderina (300 mg).
La absorcion de hierro es mejor cuando los
alimentos contienen Fe en forma de hem (carne). Diversos alimentos (p. ej.,
polifenoles y fitatos de fibras vegetales, taninos del té, fosfoproteínas,
salvado) disminuyen la absorción de Fe no hemínico. En consecuencia, muchas
interacciones entre alimentos reducen su biodisponibilidad. El ácido ascórbico
es el único elemento conocido de los alimentos habituales capaz de aumentar la
biodisponibilidad de Fe no hemínico.
Si bien la absorción real de Fe se
produce en el duodeno y el yeyuno superior, también se afecta por otras
actividades GI. El Fe no hemínico de la dieta se reduce a estado ferroso y las
secreciones gástricas lo liberan de los puntos de fijación de los alimentos. Los
mecanismos reales de absorción del Fe no se conocen aún por completo. No
obstante, las células mucosas intestinales regulan, de alguna manera importante,
la absorción. La señal principal para la célula intestinal parece guardar
relación con la reserva corporal total de Fe. Se ha comprobado que la
concentración sérica de ferritina guarda una relación inversa con la cantidad de
Fe absorbido; es posible que la ferritina (o la transferrina) proporcione dicha
señal. Una eritropoyesis aumentada (p. ej., anemia hemolítica congénita) también
puede afectar la regulación de la captación de Fe y su retención por las células
mucosas del intestino.
El Fe captado
por la célula mucosa intestinal se transfiere a la transferrina, una
proteína transportadora de Fe sintetizada en el hígado que posee dos sitios de
fijación; este sistema recoge Fe de las células (intestinales, macrófagos) y lo
cede a receptores específicos presentes en eritroblastos, células placentarias y
hepatocitos. La transferrina se une a receptores de membrana específicos en los
eritroblastos; el complejo transferrina-Fe penetra en el precursor eritrocitario
mediante endocitosis y el Fe se transfiere al interior de la mitocondria, que
introduce el Fe en la protoporfirina para convertirla en hem. La transferrina
(semivida plasmática de 8 días) es expulsada para su posterior
reutilización.
La transferrina transfiere
el Fe no utilizado en la eritropoyesis al depósito de almacenamiento, que tiene
dos formas. La más importante es la ferritina (una familia heterogénea de
proteínas formadas alrededor de un núcleo de Fe), que constituye una fracción
soluble y activa de depósito localizado en el hígado (en los hepatocitos), la
médula ósea, el bazo (en los macrófagos), los hematíes y el suero. El depósito
tisular de ferritina es muy lábil y fácilmente disponible para cualquier
requerimiento de Fe por parte del organismo. Al parecer, la ferritina circulante
(sérica) tiene su origen en el sistema mononuclear fagocítico
(reticuloendotelial) y su concentración circulante va paralela al tamaño de los
depósitos corporales (1 ng/ml corresponde a 8 mg de Fe en el depósito de
almacenamiento). El segundo depósito se localiza en la hemosiderina, un
depósito relativamente insoluble almacenado sobre todo en el hígado (en células
de Kupffer) y en la médula ósea (en macrófagos).
Dado que la absorción de Fe es tan
limitada, el organismo tiene un mecanismo muy conservador para satisfacer sus
requerimientos diarios. Las células del sistema mononuclear fagocítico fagocitan
los hematíes envejecidos. La digestión rápida proporciona Fe disponible que es
captado por la transferrina para su reutilización. Este sistema de reutilización
de Fe es tan eficaz que alrededor del 97% de las necesidades diarias de Fe (unos
25 mg) se satisfacen a partir de este depósito de almacenamiento; otro miligramo
procede de la absorción intestinal.
ANEMIA FERROPENICA
Siempre debe considerarse la hemorragia
como el principal mecanismo de ferropenia, que es la causa más frecuente de
anemia; en los adultos, la hemorragia es, virtualmente, el único mecanismo
posible. En los varones, la causa más habitual es la hemorragia crónica oculta,
por lo general del tracto GI. En las mujeres premenopáusicas, el origen puede
ser la pérdida menstrual, aunque siempre deben tenerse en cuenta otros
mecanismos. Aunque podría suponerse que la ausencia de menstruaciones durante la
gestación protege a la madre de la ferropenia, es necesario el aporte de un
suplemento de Fe debido a que se produce una pérdida neta de Fe hacia el feto en
desarrollo.
La
ferropenia también puede deberse a un aumento de los requerimientos de Fe, a una
disminución de su absorción o a ambos mecanismos. La aparición de ferropenia es
probable durante los primeros dos años de vida si el Fe de la dieta es
inadecuado para las demandas de un crecimiento rápido. Las adolescentes pueden
presentar ferropenia por dieta inadecuada, por el incremento de los
requerimientos del crecimiento y por la menstruación. El repentino crecimiento
en los varones adolescentes también puede producir un aumento significativo de
la demanda de Fe, ocasionando una eritropoyesis deficiente de Fe.
Otras causas posibles de este tipo de
anemia son la disminución de la absorción de Fe tras gastrectomía, síndromes de
malabsorción por alteraciones del intestino delgado superior y, en ocasiones,
ciertas formas de pica (principalmente yeso), pero estos mecanismos son raros en
comparación con la hemorragia. La mayoría de las formas de pica (p. ej.,
almidón, yeso, hielo) se asocian a una reducción de la ingestión por la
sustitución calórica, en vez de a una disminución de la absorción. En
situaciones de hemólisis intravascular crónica (p. ej., hemoglobinuria
paroxística nocturna, coagulación intravascular diseminada crónica, válvulas
protésicas cardíacas defectuosas), la fragmentación de los hematíes (reconocible
en una extensión de sangre periférica) puede originar carencia de Fe por
hemoglobinuria y hemosiderinuria crónicas.
El Fe se absorbe con dificultad, por lo
que la mayoría de las personas apenas satisfacen sus requerimientos diarios. Las
pérdidas añadidas por menstruación (una media de 0,5 mg/d), embarazo (0,5-0,8
mg/d), lactancia (0,4 mg/d) y hemorragias (secundarias a enfermedad, accidente o
flebotomía) provocan ferropenia con rapidez, que tiene lugar en diferentes
estadios, siendo la depleción de Fe el último de ellos.
La fisiopatologia de la Anemia por Deficit de Hierro comprende varios estadios:
Estadio 1: La pérdida de Fe supera a la ingestión,
lo que provoca un agotamiento progresivo de los depósitos de Fe (representados
por el contenido de Fe en la médula ósea). Si bien la Hb y el Fe sérico
permanecen normales, se registra una disminución de la concentración de
ferritina sérica(<20 ng/ml). A medida que se reduce el depósito de Fe, se
produce un incremento compensador en la absorción del Fe de la dieta y en la
concentración de transferrina (representado por un aumento en la capacidad de
fijación de Fe).
Estadio 2: Los depósitos agotados de Fe no pueden
satisfacer las necesidades de la médula eritroide. Mientras que el nivel de
transferrina plasmática se eleva, la concentración sérica de Fe disminuye, lo
que origina una reducción progresiva del Fe disponible para la formación de
hematíes. La eritropoyesis se altera cuando el Fe sérico disminuye por debajo de
50 mg/dl (<9 mmol/l) y la saturación de transferrina es inferior
al 16%. También se registra un aumento de la concentración del receptor de
ferritina sérica (>8,5 mg/l).
Además de las manifestaciones habituales
de anemia, aparecen algunos síntomas específicos de ferropenia. En los casos
crónicos y graves, los pacientes pueden presentar pica (p. ej., polvo, pintura)
o pagofagia (deseo de ingerir hielo), glositis, queilosis y coiloniquia y, más
raramente, disfagia asociada a una membrana esofágica poscricoidea. Finalmente,
pueden aparecer astenia y disminución de la resistencia como consecuencia de un
efecto distinto sobre los tejidos (quizá por disfunción de las enzimas celulares
que contienen Fe).
ANEMIA POR DEFICIT DE TRANSPORTE DE HIERRO
Esta anemia es extraordinariamente
infrecuente y se produce cuando el Fe no puede movilizarse desde los lugares de
depósito (p. ej., células mucosas, hígado) hacia los precursores
eritropoyéticos. El mecanismo etiológico se atribuye a la ausencia de
transferrina o a la presencia de una molécula de transferrina defectuosa. Además
de la anemia, es característica la hemosiderosis del tejido linfoide,
especialmente en el tracto GI.
ANEMIA POR DEFICIT EN LA UTILIZACION DEL HIERRO
Esta clase de anemias se
deben a la utilización inadecuada o anómala del Fe intracelular para la síntesis
de Hb, a pesar de la existencia de unas cantidades adecuadas o aumentadas de Fe
en el interior de las mitocondrias de las células precursoras de los hematíes.
Este defecto incluye las hemoglobinopatías, sobre todo de tipo
talasémico, y la anemia sideroblástica o mielodisplásica. Debido a que
existen otras características clínicas y de laboratorio que contribuyen a
definir las talasemias, el término sideroblástico suele aplicarse al segundo
subgrupo. En la actualidad, los estados sideroblásticos primarios (o
idiopáticos) se incluyen habitualmente en el síndrome mielodisplásico, por lo
que algunos autores utilizan el término sideroblastosis para expresar que todas
las formas son, en realidad, displásicas.
Aunque la anemia sideroblástica es
frecuentemente microcítica e hipocrómica, existe una amplitud de distribución de
volumen eritrocitario (ADE) elevada como resultado de la población dimórfica
(pequeño y gran tamaño) de células circulantes; la heterogeneidad celular se
reconoce en el examen de la extensión de sangre periférica. Una clave importante
indicativa de síntesis defectuosa del grupo hem en la sangre periférica es la
presencia de hematíes en diana con punteado policromatófilo (es decir,
siderocitos). Otro dato de laboratorio es el aumento en las concentraciones de
Fe y ferritina séricos y en la saturación de transferrina. En la médula ósea
existe una hiperplasia eritroide con características displásicas; la tinción por
el Fe revela el rasgo morfológico patognomónico, consistente en la presencia de
mitocondrias paranucleares grandes cargadas de Fe en los eritroblastos
(sideroblastos en anillo). En las formas adquiridas, especialmente en la forma
primaria o idiopática, son evidentes otras características de mielodisplasia
como la granulopoyesis defectuosa o la existencia de megacariocitos
uninucleados.
Estas anemias se caracterizan por una
eritropoyesis ineficaz, que se define clínicamente como anemia y
reticulocitopenia absoluta o relativa en presencia de hiperplasia eritroide. El
Fe marcado con radioisótopos se transfiere rápidamente de la transferrina
plasmática a la médula ósea, pero no puede reaparecer en los hematíes
circulantes a un ritmo normal. Los estudios ferrocinéticos proporcionan datos
sugestivos de eritropoyesis ineficaz, lo que implica que la maduración eritroide
anómala provoca un aumento de la destrucción intramedular de los
hematíes.
Se desconocen los mecanismos
fisiopatológicos específicos que producen sideroblastos reconocibles. El número
de enfermedades que se asocian en ocasiones a sideroblastosis es muy extenso y
casi todas ellas originan, en general, otros defectos más típicos en la
eritropoyesis.
La anemia sideroblástica pura sin
modificaciones en la estructura y la producción de leucocitos y plaquetas es
extremadamente rara. Prácticamente todos los casos en que se constatan estos
cambios se asocian a un estado mielodisplásico.
ANEMIA DE LAS ENFERMEDADES CRONICAS
(Anemia por déficit en la reutilización
de hierro)
Este tipo de anemia representa la segunda
forma de anemia más frecuente en el mundo. En fases iniciales, los hematíes son
normocíticos y, con el paso del tiempo, se vuelven microcíticos. La
característica más importante es que la masa eritroide medular no se expande de
manera adecuada en respuesta a la anemia.
Se creía que esta anemia se presentaba
como parte de ciertas enfermedades crónicas, entre las cuales las identificadas
con mayor frecuencia son las infecciones, las enfermedades inflamatorias (en
particular AR) y las neoplasias; sin embargo, la enfermedad subyacente no es
necesariamente crónica, dado que las características fisiopatológicas de dicha
anemia aparecen de forma transitoria prácticamente durante cualquier infección o
inflamación. Se han identificado tres mecanismos fisiopatológicos: 1) se ha
demostrado un acortamiento discreto en la supervivencia de los hematíes (en el
contexto de una producción compensadora para una médula ósea normal) en
pacientes con neoplasias y enfermedades infecciosas granulomatosas crónicas. El
mecanismo exacto de esta lesión extracorpuscular de los hematíes se desconoce,
si bien se ha hallado recientemente una proteína de 50.000 kD en algunos
pacientes con cáncer. 2) La síntesis de EPO y la capacidad de respuesta de la
médula ósea están disminuidas, lo que ocasiona una eritropoyesis deficiente. Las
citocinas producidas por macrófagos (p. ej., interleucina-1b, factor de necrosis
tumoral-a, interferón-b), halladas en pacientes
con infecciones, estados inflamatorios y neoplasias, provocan esta disminución
en la producción de EPO. 3) El metabolismo intracelular del Fe está alterado. El
reciclaje eficaz del Fe procedente de los hematíes envejecidos es fundamental
para mantener su equilibrio. En las enfermedades crónicas, las células
reticulares retienen con tenacidad el Fe de los hematíes viejos, impidiendo que
la eritrona disponga de éste para la síntesis de Hb. Existen reticulocitopenia e
incapacidad para compensar la anemia mediante hiperplasia eritroide. La
alteración del metabolismo del Fe y la eritropoyesis deficiente resultante
también son consecuencia de la producción de citocinas inflamatorias.
c. ANEMIAS NORMOCITICAS
En estos casos el volumen corpuscular medio está dentro de lo normal en el rango de referencia.
Ejemplos de este tipo de anemias son:
- Las anemias producidas por enfermedades crónicas (casi siempre).
- Las anemias hemolíticas (salvo reticulocitosis).
- Aplasia medular (la mayoría).
- Pérdidas agudas (salvo infrecuente reticulocitosis).
NORMOCITICAS NORMOCROMICAS
ANEMIAS HIPOPROLIFERATIVAS
El mecanismo fisiopatológico de las
anemias hipoproliferativas parece ser una reducción, relativa o absoluta, de la
síntesis de EPO o un estado hipometabólico con insuficiente respuesta a la EPO.
Como ya se mencionó, la anemia ferropénica y la anemia de las enfermedades
crónicas son cuadros hipoproliferativos, dado que cursan con hiperplasia
eritroide limitada y con disminución de la producción de EPO y de la capacidad
de respuesta de la médula ósea. La hipoproliferación se asocia frecuentemente a
anemias de origen renal o a estados hipometabólicos (p. ej., hipotiroidismo,
hipopituitarismo) y de deprivación proteica, que ocasionan una reducción en la
síntesis de EPO.
La gravedad de la anemia se correlaciona
con la intensidad de la disfunción renal. La producción renal de EPO es
paralela, en general, a la función excretora del riñón, de manera que la anemia
aparece cuando el aclaramiento de creatinina es menor de 45 ml/min. La
disminución de la eritropoyesis provocada por la reducción de EPO se expresa
como reticulocitopenia periférica y una respuesta medular inferior a la normal
(ausencia de hiperplasia eritroide para el grado de anemia). Las lesiones
renales que afectan principalmente a la región glomerular (p. ej., amiloidosis,
nefropatía diabética) suelen ocasionar la anemia más grave para el grado de
insuficiencia excretora.
El término anemia de la insuficiencia
renal sólo hace referencia al mecanismo hipoproliferativo
hipoeritropoyetinémico, si bien otros mecanismos pueden complicar la gravedad
del trastorno. En la uremia es frecuente un componente hemolítico leve; su
origen se desconoce, pero se relaciona con la retención de los "residuos
metabólicos de la uremia" que, de alguna forma, lesionan los hematíes. Menos
común, pero más fácilmente reconocible, es la anemia asociada a fragmentación de
los hematíes (anemia hemolítica microangiopática) que surge cuando se lesiona el
endotelio vascular renal (p. ej., en la hipertensión maligna, la poliarteritis
nodosa o la necrosis cortical aguda). La hemólisis microangiopática puede
reconocerse en la extensión de la sangre periférica por la fragmentación de los
hematíes con trombocitopenia asociada habitualmente. En los niños puede tratarse
de una enfermedad aguda, a menudo mortal, denominada síndrome hemolítico-urémico
ANEMIA APLASICA
Anemia por pérdida de precursores de
hematíes, ya sea por un defecto en la reserva de células madres o por una lesión
del microambiente que mantiene a la médula ósea, y que cursa frecuentemente con
valores de VCM en el límite superior. El término anemia
aplásica suele implicar una panhipoplasia de la médula con leucopenia y
trombocitopenia asociadas. Esta confusión de la nomenclatura ha conducido al
término de aplasia eritrocitaria pura, que define la reducción selectiva
pronunciada o la ausencia de precursores eritroides. Si bien ambas enfermedades
son poco habituales, la anemia aplásica se observa con mayor frecuencia.
Aproximadamente el 50% de
los casos de anemia aplásica verdadera son idiopáticos y ocurren más a menudo en
adolescentes y adultos jóvenes. Las causas reconocidas son agentes químicos (p.
ej., benceno, arsénico inorgánico), radiaciones y fármacos (p. ej.,
antineoplásicos, antibióticos, AINE, anticonvulsivantes). El mecanismo de estos
trastornos se desconoce, aunque parece existir una hipersensibilidad selectiva
(quizá genética). Una forma muy rara de anemia aplásica, la anemia de Fanconi
(un tipo de anemia aplásica familiar con anomalías óseas, microcefalia,
hipogenitalismo y pigmentación parda de la piel), aparece en niños con
cromosomas anómalos. Como consecuencia, el diagnóstico específico no suele
realizarse hasta que sobreviene alguna enfermedad, sobre todo infecciones agudas
o trastornos inflamatorios, que puede provocar citopenias periféricas. Cuando se
resuelve la enfermedad intercurrente, los valores periféricos recuperan la
normalidad a pesar de la masa medular reducida.
La aplasia
eritrocitaria pura implica un mecanismo selectivamente destructor de los
precursores eritroides. La eritroblastopenia aguda es una desaparición
reversible y breve de los precursores eritrocitarios de la médula que puede
aparecer en el curso de diversas enfermedades víricas agudas, especialmente en
niños. La infección por parvovirus humano parece ser la causa más frecuente de
estos episodios. De hecho, puede reconocerse fortuitamente, dado que la anemia
perdura más tiempo que la infección aguda. La aplasia eritrocitaria crónica se
ha asociado a trastornos hemolíticos (eritroblastopenia aguda), timomas,
trastornos inmunológicos y, con menor frecuencia, a fármacos (p. ej.,
tranquilizantes, anticonvulsivantes), tóxicos (fosfatos orgánicos), déficit de
riboflavina y leucemia linfática crónica. Se han descrito casos en la edad
adulta de una forma congénita rara, el síndrome de Blackfan-Diamond,
aunque en principio se creía que sólo se manifestaba en la infancia. La
presencia de anomalías óseas en los pulgares y dedos de las manos y la talla
corta sugieren el diagnóstico.
MIELODISPLASIA
La anemia suele ser una característica
destacada de la mielodisplasia (v. cap. 138  ). La anemia es
normocrómica, normocítica y se asocia a una disminución de la actividad
eritroide en la médula ósea, a alteraciones megaloblastoides y displásicas y, en
ocasiones, a un aumento del número de sideroblastos en anillo, como ya se
mencionó. La anemia sintomática suele controlarse mediante el tratamiento con
EPO, que es particularmente útil en los pacientes con niveles séricos de EPO
inferiores a los esperados para su grado de anemia. Se requieren dosis
farmacológicas de EPO, ya que existe una eritropoyesis deficiente y que la
anemia no se debe a un descenso de su secreción; aproximadamente el 50% de los
pacientes responden, con lo que se elimina la necesidad de transfusiones.
). La anemia es
normocrómica, normocítica y se asocia a una disminución de la actividad
eritroide en la médula ósea, a alteraciones megaloblastoides y displásicas y, en
ocasiones, a un aumento del número de sideroblastos en anillo, como ya se
mencionó. La anemia sintomática suele controlarse mediante el tratamiento con
EPO, que es particularmente útil en los pacientes con niveles séricos de EPO
inferiores a los esperados para su grado de anemia. Se requieren dosis
farmacológicas de EPO, ya que existe una eritropoyesis deficiente y que la
anemia no se debe a un descenso de su secreción; aproximadamente el 50% de los
pacientes responden, con lo que se elimina la necesidad de transfusiones.
ANEMIA POST HEMORRAGICA AGUDA
La reserva medular es limitada, por lo
que la anemia puede ser el resultado de cualquier hemorragia masiva, debida a la
rotura o la incisión traumática o espontánea de un vaso sanguíneo de gran
calibre, a la erosión de una arteria por lesiones (p. ej., úlcera péptica,
neoplasias) o al fracaso de los mecanismos hemostáticos normales. Los efectos
inmediatos dependen de la duración y el volumen de la hemorragia. La pérdida
súbita de 1/3 del volumen sanguíneo puede resultar mortal, pero pueden perderse
lentamente hasta 2/3 durante 24 h sin este riesgo. Los síntomas se deben al
descenso repentino del volumen sanguíneo y a la consiguiente hemodilución, con
una disminución en la capacidad transportadora de O2 de la
sangre.
El carácter de la hemorragia determina la
intensidad de los síntomas. Pueden aparecer vahídos, vértigos, sed, sudación,
pulso débil y rápido y respiración rápida (inicialmente profunda y luego
superficial). La hipotensión ortostática es frecuente. La PA puede elevarse
ligeramente al inicio debido a la vasoconstricción arteriolar refleja, pero
luego disminuye de forma gradual. Si continúa la hemorragia, la PA puede
descender y producir la muerte del paciente.
Durante e inmediatamente después de la
hemorragia, el recuento de hematíes, la Hb y el Hto pueden encontrarse
engañosamente elevados como consecuencia de la vasoconstricción. A las pocas
horas, los líquidos tisulares penetran en la circulación, produciendo
hemodilución y un descenso del recuento de hematíes y de Hb proporcional a la
gravedad de la hemorragia. La anemia resultante es normocítica. En las primeras
horas puede aparecer granulocitosis polimorfonuclear y elevación del recuento
plaquetario. Varios días después del episodio hemorrágico aparecen signos de
regeneración (es decir, reticulocitosis): las extensiones sanguíneas pueden
revelar policromatofilia y macrocitosis ligera; si la hemorragia fue masiva y
aguda pueden verse algunos normoblastos y leucocitos inmaduros.
ANEMIA HEMOLITICA
Al final de su vida media normal
(alrededor de 120 d), los hematíes se eliminan por los componentes del sistema
mononuclear fagocítico, sobre todo en el bazo, donde tiene lugar el catabolismo
de la Hb. El rasgo característico de la hemólisis es el acortamiento de la vida
media de los hematíes. La anemia hemolítica aparece cuando la producción de
hematíes en la médula ósea es incapaz de compensar esta supervivencia
acortada.
La mayoría de los procesos
de hemólisis son extravasculares, es decir, se producen en las células
fagocíticas del bazo, el hígado y la médula ósea. La hemólisis tiene su origen
1) en anomalías intrínsecas del contenido de los hematíes (Hb o enzimas) o de su
membrana (permeabilidad, estructura o contenido lipídico), o bien 2) en
problemas extrínsecos a los hematíes (anticuerpos séricos, traumatismos en la
circulación o microorganismos infecciosos). El bazo suele intervenir, de manera
que reduce la supervivencia de los hematíes al destruir los que son ligeramente
anómalos o están cubiertos con anticuerpos calientes. En caso de existir
esplenomegalia, puede haber atrapamiento (secuestro) incluso de hematíes
normales. Los hematíes muy alterados o los recubiertos de anticuerpos fríos o
complemento (C3) se destruyen en la circulación o en el hígado, órgano capaz de
eliminar con eficacia las células lesionadas gracias a su elevado flujo
sanguíneo.
La hemólisis
intravascular es infrecuente y cursa con hemoglobinuria cuando la Hb
liberada en el plasma supera la capacidad fijadora de Hb de las proteínas
fijadoras del plasma (p. ej., haptoglobina). La Hb se reabsorbe en las células
de los túbulos renales, donde el Fe se transforma en hemosiderina, una parte de
la cual se asimila para reutilizarse y otra parte llega a la orina cuando se
desprenden las células a la luz tubular. La identificación de hemosiderinuria en
una muestra de orina reciente demuestra la existencia de un proceso hemolítico
intravascular.
Las manifestaciones sistémicas son
parecidas a las que se observan en otros tipos de anemia. La hemólisis puede ser
aguda, crónica o episódica. Las crisis hemolíticas (hemólisis aguda grave) son
infrecuentes y pueden acompañarse de fiebre, escalofríos, dorsalgia, dolor
abdominal, postración y shock. En los casos graves, aumenta la hemólisis
(ictericia, esplenomegalia y, en ciertos tipos de hemólisis, hemoglobinuria y
hemosiderinuria) y la eritropoyesis (reticulocitosis e hiperactividad de la
médula ósea). En los estados hemolíticos crónicos, la anemia puede empeorar por
la aparición de crisis aplásicas (fracaso temporal de la eritropoyesis), que
suelen relacionarse con infecciones, a menudo por parvovirus.
En la hemólisis causada por defectos
extrínsecos a los hematíes no puede identificarse ni implicarse ninguna anomalía
de los hematíes; su destrucción se debe a circunstancias ajenas a los propios
hematíes. Las células de donantes se destruyen a la misma velocidad que las
células autólogas.
ANEMIA HEMOLITICA AUTOINMUNE
La anemia hemolítica autoinmune (AHAI) se
identifica por la presencia de autoanticuerpos que reaccionan con los hematíes.
Estos anticuerpos se detectan mediante la prueba de la antiglobulina (Coombs)
directa. En ella, se añade suero antiglobulina a hematíes lavados procedentes
del paciente; la aglutinación indica la existencia de inmunoglobulinas o
componentes del complemento unidos a los hematíes. Por otra parte, la mezcla del
plasma del paciente con hematíes normales detecta la presencia de tales
anticuerpos (libres) en el plasma (prueba de la antiglobulina [Coombs]
indirecta). En general, la intensidad de la prueba de la antiglobulina directa
se correlaciona con el número de moléculas de IgG o C3 unidas a los hematíes y,
aunque no con una relación exacta, con la tasa de hemólisis. Una prueba de
antiglobulina indirecta positiva (p. ej., presencia de anticuerpos libres
antihematíes) en ausencia de una prueba directa positiva no indica la existencia
de una hemólisis de origen inmunitario, sino que suele definir un aloanticuerpo
producido por embarazo, transfusiones anteriores o reactividad cruzada con
lectinas. La identificación de anticuerpos calientes tampoco define la
hemólisis, ya que 1/10.000 donantes de sangre normales tienen resultados
positivos en esta prueba.
ANEMIA HEMOLITICA TRAUMATICA
(Anemia hemolítica
microangiopática)
Cuando los hematíes se exponen a una
fricción o a una turbulencia excesiva en la circulación, aparecen fragmentos
eritrocitarios de formas extrañas (p. ej., formas en triángulo o casco) en la
sangre periférica que permiten efectuar el diagnóstico. Debido a estos
fragmentos, el VCM puede ser bajo y la ADE, que refleja la anisocitosis,
elevada. El traumatismo puede originarse 1) en el exterior del vaso, como en la
hemoglobinuria de la marcha, del karate o de los músicos de bongos; 2) en el
interior del corazón, como en la estenosis aórtica calcificada y en prótesis
valvulares aórticas defectuosas; 3) en arteriolas, como en la hipertensión grave
(sobre todo maligna), en algunos tumores malignos y en la poliarteritis nudosa,
o 4) en arteriolas terminales, como en la púrpura trombocitopénica trombótica y
en la coagulación intravascular diseminada. En este último caso aparecen
deficiencias en los factores de la coagulación. El tratamiento se dirige al proceso
subyacente. En ocasiones se sobreañade una anemia ferropénica a la hemólisis
como consecuencia de una hemosiderinuria crónica, la cual puede responder al
tratamiento con Fe.
ANEMIA HEMOLITICA INFECCIOSA
Los agentes infecciosos pueden producir
anemia hemolítica por la acción directa de toxinas (p. ej., Clostridium
perfringens, estreptococos hemolíticos a o b o meningococos) o por
la invasión y destrucción de los hematíes por el propio microorganismo (p. ej.,
Plasmodium y Bartonella sp).
ANEMIA POR DEFECTOS INTRINSECOS DE LOS HEMATIES
ANEMIA POR ALTERACIONES DE MEMBRANA ERITROCITARIA
El análisis del citoesqueleto de la
membrana eritrocitaria revela que la mayoría de las alteraciones estructurales
hereditarias o adquiridas se deben a modificaciones en las proteínas de
membrana. Los estudios realizados sobre estas proteínas citoesqueléticas
(a-espectrina, b-espectrina, proteína
4.1, F-actina, anquirina) han demostrado la existencia de anomalías
cuantitativas y funcionales en estas anemias hemolíticas. A menudo se observa un
patrón familiar en los casos congénitos. No obstante, se desconoce el mecanismo
por el que estas alteraciones de las proteínas estructurales provocan
hemólisis.
La esferocitosis
hereditaria (ictericia crónica familiar, ictericia hemolítica congénita,
ictericia acolúrica crónica, esferocitosis familiar, anemia esferocítica) es una
enfermedad crónica que se hereda como rasgo dominante y que se caracteriza por
hemólisis de hematíes esferoidales, anemia, ictericia y esplenomegalia. Aunque
en general uno o más miembros de la familia presentan ictericia, anemia o
esplenomegalia, una o más generaciones pueden estar libres debido a las
variaciones en el grado de penetrancia del gen.
La eliptocitosis
hereditaria (ovalocitosis) es un raro trastorno de herencia autosómica
dominante en el que los hematíes son ovalados o elípticos. Generalmente no
existe hemólisis o es escasa, con anemia leve o ausente y, a menudo,
esplenomegalia. La anomalía eritrocitaria parece deberse a una alteración en las
proteínas de la membrana.
En la esferocitosis hereditaria, la
superficie de la membrana celular está reducida de forma desproporcionada al
contenido intracelular. Diversas anomalías proteicas de la membrana
eritrocitaria provocan cambios esferocíticos. La disminución de la superficie de
las células impide la flexibilidad necesaria para atravesar la microcirculación
esplénica. Como consecuencia, la hemólisis tiene lugar en el bazo.
Los síntomas y signos de
la esferocitosis hereditaria suelen ser leves y la anemia puede estar tan
bien compensada que no se reconozca hasta que una enfermedad intercurrente
suprima la eritropoyesis. En casos graves puede haber ictericia moderada y
síntomas de anemia. Pueden presentarse crisis aplásicas debidas a infecciones
intercurrentes que, en ocasiones, exacerban la anemia. La esplenomegalia es casi
invariable, pero raras veces causa molestias abdominales. Puede existir
hepatomegalia y es frecuente la colelitiasis (cálculos pigmentarios), que puede
ser el síntoma cardinal de presentación. Ocasionalmente se observan alteraciones
esqueléticas congénitas (p. ej., turricefalia y polidactilia).
Las características
clínicas de la eliptocitosis hereditaria son similares a las que se
observan en la esferocitosis hereditaria, pero tienden a ser más leves.
TRASTORNOS ADQUIRIDOS DE LA MEMBRANA ERITROCITARIA
La estomatocitosis
es un trastorno de los hematíes en los que un patrón en forma de boca o
hendidura sustituye la zona de palidez central normal. Estas células aparecen en
anemias hemolíticas congénitas y adquiridas. Los síntomas se relacionan
directamente con el grado de anemia.
La rara forma
congénita, que se hereda de forma autosómica, es la mejor caracterizada. La
membrana eritrocitaria es hiperpermeable a los cationes monovalentes, mientras
que el movimiento de los cationes divalentes y de los aniones es normal. Los
hematíes circulantes son estomatocíticos (20-30%); la fragilidad eritrocitaria
está elevada, al igual que la autohemólisis, que se corrige de forma inconstante
con glucosa. La esplenectomía produce, en algunos casos, la mejoría de la
anemia.
La estomatocitosis
adquirida con anemia hemolítica aparece principalmente en los abusos
recientes de alcohol. Los estomatocitos de la sangre periférica y la hemólisis
acelerada desaparecen a las 2 sem de la retirada del alcohol.
Anemia debida a hipofosfatemia. La deformabilidad
de los hematíes depende de las concentraciones intracelulares de ATP, Ca y Mg.
Como el contenido eritrocitario de ATP se relaciona con la concentración sérica
de P, la hipofosfatemia (valores séricos <0,5 mg/dl [<0,16 mmol/l])
provoca una depleción eritrocitaria de ATP; las complejas secuelas metabólicas
de la hipofosfatemia también incluyen depleción de 2,3-difosfoglicerato,
desviación a la izquierda de la curva de disociación del O2,
disminución de la utilización de glucosa y producción de lactato. Los hematíes
resultantes son rígidos, no flexibles y susceptibles de lesionarse en el lecho
circulatorio capilar, lo que origina una anemia hemolítica con alteración de la
membrana y microsferocitosis.
Puede producirse una hipofosfatemia grave
en el síndrome de abstinencia alcohólica, la diabetes mellitus, la fase de
recuperación (diurética) tras quemaduras graves, la hiperalimentación, la
alcalosis respiratoria grave o en pacientes urémicos sometidos a diálisis y
tratados con antiácidos. Dado que estos cambios pueden prevenirse o corregirse
si se mantiene el ATP celular con suplementos de fosfato, el tratamiento debe
consistir en la protección contra la hipofosfatemia cuando se prevea dicha
situación y en la administración de fosfato cuando se advierta depleción.
HEMOGLOBINOPATIAS O ANEMIA POR DEFECTOS DE LA HEMOGLOBINA
(Hemoglobinopatías)
Alteraciones genéticas de la molécula
de Hb que se demuestran por cambios en las características químicas, en la
movilidad electroforética o en otras propiedades físicas. La molécula de Hb normal en el adulto (Hb
A) consta de dos pares de cadenas polipeptídicas denominadas a y b. La Hb fetal (Hb F, en
la que las cadenas g sustituyen a las cadenas b) disminuye gradualmente
en los primeros meses de vida hasta que representa menos del 2% de la Hb total
del adulto. En ciertos trastornos
de la síntesis de Hb y en los estados aplásicos y mieloproliferativos, la Hb F
puede estar aumentada. La sangre normal también contiene, como máximo, un 2,5%
de Hb A2 (compuesta de cadenas a y d).
Los tipos de cadenas y la estructura
química de sus polipéptidos están controlados genéticamente. Pueden aparecer
defectos en las moléculas de Hb, con propiedades físicas o químicas anómalas;
algunos provocan anemias que son graves en los individuos homocigotos, pero
leves en los portadores heterocigotos. Algunas personas pueden ser heterocigotas
para dos de estas anomalías y presentar una anemia con caracteríticas de ambos
rasgos.
Las Hb anómalas, diferenciadas por su
movilidad electroforética, se designan alfabéticamente en orden de
descubrimiento (p. ej., A, B, C), si bien la primera de ellas, la Hb de las
células falciformes, se denominó Hb S. Las Hb estructuralmente diferentes con la
misma movilidad electroforética también se denominan según la ciudad donde se
descubrieron (p. ej., Hb S Memphis, Hb C Harlem). En Estados Unidos, las
hemoglobinopatías importantes se deben a la síntesis defectuosa de Hb S y Hb C y
a las talasemias; la inmigración de personas procedentes del sudeste asiático ha
conducido al reconocimiento frecuente de la Hb E en la práctica clínica. Por
tradición de laboratorio, la Hb electroforética de concentración superior se
nombra en primer lugar (p. ej., AS en el rasgo de células falciformes, mientras
que en SA [anemia de células falciformes asociada a talasemia b] la
concentración de Hb A está reducida por la presencia de la talasemia y la Hb S).
Son:
- Anemia de celulas falciformes
- Hemoglobinopatia C
- Hemoglobinopatia S-C
- Hemoglobinopatia E
- Talasemia
ANEMIA DE CELULAS FALCIFORMES
Anemia hemolítica crónica que se
presenta casi exclusivamente en individuos de raza negra, caracterizada por
hematíes falciformes, debido a la herencia homocigota de Hb S.
Los homocigotos presentan anemia de
células falciformes (aproximadamente el 0,3% de los individuos de raza negra de
Estados Unidos); los heterocigotos (8-13% de los negros) no son anémicos, pero
puede demostrarse el rasgo falciforme (falcemia) in vitro.
En la Hb S, la valina se sustituye por
ácido glutámico en el sexto aminoácido de la cadena b. Este cambio reduce su
carga eléctrica y determina que se mueva con más lentitud que la Hb A hacia el
ánodo en el análisis electroforético. La desoxi-Hb S es mucho menos soluble que
la desoxi-Hb A; forma un gel semisólido (polimerización) de tactoides con forma
de bastoncillo, lo que ocasiona que los hematíes adopten una forma de hoz en las
zonas con Po2 baja. Los hematíes deformados y rígidos se adhieren al
endotelio vascular y taponan las pequeñas arteriolas y capilares, lo que
conlleva oclusión e infarto. Como los hematíes falciformes son demasiado
frágiles para resistir el traumatismo mecánico de la circulación, se produce su
hemólisis cuando se introducen en el torrente circulatorio.
En homocigotos, las
manifestaciones clínicas se deben tanto a la anemia como a los episodios
vasooclusivos que ocasionan isquemia tisular e infarto. El crecimiento y el
desarrollo están alterados y se incrementa la susceptibilidad a las infecciones.
La anemia, en general grave, es muy variable de un paciente a otro; la mayoría
presenta ictericia leve (nivel de bilirrubina de 2-4 mg/dl [34-68 mmol/l]). La
anemia puede exacerbarse en los niños por secuestro agudo de células falciformes
en el bazo.
Los pacientes pueden presentar un
desarrollo deficiente y suelen tener un tronco relativamente corto con
extremidades largas y un cráneo en torre. La hiperactividad crónica de la médula
produce unas alteraciones óseas típicas, detectables en las radiografías: son
característicos el ensanchamiento del díploe de los huesos del cráneo y el
aspecto en rayos de sol de las trabeculaciones diploicas. Los huesos largos
muestran a menudo engrosamiento cortical, densidades irregulares y neoformación
ósea en el interior del canal medular. En niños es frecuente la
hepatosplenomegalia, pero como resultado de los infartos repetidos y la
consiguiente fibrosis, el bazo suele ser muy pequeño en los adultos debido a
esta "autoesplenectomía". Por esta razón, un bazo palpable en estos pacientes
sugiere que la Hb es de tipo S-C o S-A. La cardiomegalia es habitual, con un
cono pulmonar prominente. Los ruidos cardíacos pueden simular una cardiopatía
reumática o congénita. Es frecuente la colelitiasis.
Las crisis aplásicas ocurren cuando la
eritropoyesis medular disminuye durante infecciones agudas (especialmente
víricas). Los infartos óseos provocan crisis dolorosas, el complejo sintomático
más común en los estados de Hb S-S, S-A y S-C. El dolor en huesos largos (p.
ej., pretibial) es la queja más frecuente; en los niños es habitual y típico un
dolor intenso en manos y pies (p. ej., síndrome de manos y pies). Pueden
aparecer artralgias con fiebre y es frecuente la necrosis avascular de la cabeza
femoral. También suelen observarse úlceras sobreelevadas crónicas en la región
de los tobillos. Los episodios de dolor abdominal intenso con vómitos pueden
simular trastornos abdominales graves; estas crisis dolorosas se asocian
generalmente a dolor de espalda y articular. La hemiplejía, la parálisis de los
pares craneales y otras alteraciones neurológicas pueden deberse a oclusión de
los principales vasos intracraneales. Las infecciones, en particular las
neumocócicas, son frecuentes, sobre todo en los primeros años de la infancia, y
conllevan una elevada tasa de mortalidad.
El síndrome torácico agudo es la
principal causa de fallecimiento en los pacientes mayores de 5 años. Se observa
en todos los grupos de edad, pero su frecuencia disminuye en la edad adulta y se
caracteriza por la aparición brusca de fiebre, dolor torácico, leucocitosis e
infiltrados en el parénquima pulmonar en la radiografía de tórax. Los
infiltrados comienzan en los lóbulos inferiores, son bilaterales en la tercera
parte de los casos y pueden asociarse a derrame pleural. Este síndrome remeda
una neumonía bacteriana y puede surgir tras dicha infección. Las lesiones se
deben a una oclusión microvascular, de manera que puede aparecer una hipoxemia
rápida. Es fundamental el apoyo ventilatorio y la consideración de la
exanguinotransfusión (para Po2 <70 mm Hg con oxigenoterapia). En
pacientes ancianos pueden verse alteraciones progresivas de las funciones
pulmonar y renal. El priapismo, una complicación grave con riesgo de desarrollar
impotencia, es más habitual en adultos jóvenes.
Los individuos
heterocigotos (Hb AS) son normales y no experimentan hemólisis, crisis
dolorosas ni complicaciones trombóticas. La incidencia de rabdomiólisis y muerte
súbita puede aumentar en pacientes con rasgo AS que practican ejercicio
constante y agotador. Es común la hipostenuria. A veces se produce una hematuria
unilateral (de mecanismo desconocido y, por lo general, procedente del riñón
izquierdo), que es autolimitada; el reconocimiento del estado falciforme
heterocigoto debe explicar la hemorragia unilateral, evitando en consecuencia
una nefrectomía innecesaria. La necrosis papilar renal típica también es más
frecuente en la anemia de células falciformes.
HEMOGLOBINOPATIA C
El grado de anemia es variable, pero
puede ser moderadamente grave. El 2-3% de los individuos de raza negra en
Estados Unidos presentan el rasgo. Los síntomas en los homocigotos se deben a la
anemia. Las artralgias son habituales. Puede haber dolor abdominal, pero no se
producen las crisis abdominales de la anemia falciforme. Los pacientes pueden
estar moderadamente ictéricos. El bazo suele estar agrandado. Pueden producirse
episodios de secuestro esplénico con dolor en el cuadrante superior izquierdo y
descensos súbitos del recuento de hematíes; los casos graves pueden requerir
esplenectomía.
En los homocigotos, la anemia es
normocítica, con un 30-100% de células en diana, esferocitos asociados y, raras
veces, hematíes con cristales en su interior, observables en la extensión. Los
pacientes con microcitosis que no son ferropénicos padecen una talasemia
a
concomitante. El número de reticulocitos está ligeramente aumentado y pueden
observarse hematíes nucleados. Los hematíes no son falciformes. La
electroforesis demuestra que toda la Hb es del tipo C. La bilirrubina sérica
está discretamente elevada y el urobilinógeno está aumentado en heces y en
orina. No existe tratamiento específico. La anemia raramente es tan grave como
para requerir transfusión sanguínea.
Los heterocigotos no suelen tener anemia
y el único hallazgo es la presencia de hematíes con diana central.
HEMOGLOBINOPATIA S-C
Dado que el 10% de los individuos de raza
negra son portadores del rasgo Hb S, la incidencia de la combinación S-C
heterocigota es mucho mayor que la de la hemoglobinopatía C en homocigosis.
Muchos casos de anemia en pacientes con falcemia pueden representar ejemplos no
detectados de hemoglobinopatía S-C. La anemia de la hemoglobinopatía S-C es
similar a la de la hemoglobinopatía C, pero más leve; algunos pacientes tienen
incluso niveles normales de Hb. La mayoría de los síntomas corresponden a los de
la anemia de células falciformes, pero generalmente son menos frecuentes y menos
graves. No obstante, a menudo hay hematuria macroscópica, hemorragias retinianas
y necrosis aséptica de la cabeza femoral. Las extensiones de sangre teñidas
muestran células en diana y células falciformes extrañas. Todas las células
presentan falciformación en preparaciones adecuadas para ello.
D. HEMOGLOBINOPATIA E
La Hb E (a2b226glu®lis) es la tercera Hb más prevalente en el mundo
(después de la A y la S), principalmente en el sudeste asiático (>15%) y en
poblaciones negras, pero es rara en los chinos.
En los heterocigotos (Hb AE) no se
encuentran alteraciones en la sangre periférica. La electroforesis de la Hb
revela aproximadamente un 30% de Hb E (hallada cerca del origen donde se
encuentran la A2, la C y la OArab) y un 70% de Hb A. En la
electroforesis en gel de agar a pH ácido, la Hb E migra junto con la A,
separándola así de la C y la OArab. El porcentaje relativo de Hb E
disminuye en asociación con talasemia a o en presencia de
ferropenia. La hemoglobinopatía E homocigota se asocia a una anemia microcítica
leve con células en diana evidentes. Los heterocigotos dobles para la Hb E y la
talasemia b presentan una enfermedad hemolítica más grave que la
hemoglobinopatía S-talasemia.
TALASEMIA
(Anemia mediterránea, leptocitosis
hereditaria, talasemias mayor y menor)
Grupo de anemias microcíticas,
crónicas y hereditarias que se caracterizan por la síntesis defectuosa de Hb y
por eritropoyesis ineficaz; son especialmente frecuentes en personas de origen
mediterráneo, africano y del sudeste asiático.
La talasemia se encuentra entre los
trastornos hemolíticos hereditarios más frecuentes. Es el resultado de la
síntesis desequilibrada de Hb, debida a una disminución en la producción de, al
menos, una cadena polipeptídica globínica (b, a, g, d).
La talasemia
b se debe a la reducción de la síntesis de cadenas
polipeptídicas b. La enfermedad es de herencia autosómica dominante: los
heterocigotos son portadores y presentan una anemia microcítica asintomática de
intesidad leve a moderada (talasemia menor); los síntomas típicos
aparecen en homocigotos (talasemia mayor). La talasemia
a,
resultante de la disminución de la producción de cadenas a, tiene un patrón
hereditario más complejo, ya que el control genético de la síntesis de las
cadenas a afecta a dos pares de genes estructurales. Los
heterocigotos para un defecto genético único (talasemia a2
[silente]) generalmente no presentan trastornos clínicos. Los heterocigotos
para un defecto genético doble o los homocigotos para un defecto genético único
(talasemia a1
[rasgo]) tienden a manifestar un cuadro clínico similar al de los
heterocigotos para la talasemia b. La herencia, tanto de un defecto genético único
como de un defecto genético doble, tiene como resultado una alteración más grave
de la síntesis de cadenas a. El déficit de cadenas a determina la formación
de tetrámeros con un exceso de cadenas b
(Hb H) o, en la infancia, de
cadenas g (Hb de Bart). El estado homocigoto para el defecto
genético doble es mortal, ya que la Hb que carece de cadenas a no transporta
O2. En la raza negra, la frecuencia genética de talasemia
a se
aproxima al 25%, observándose la expresión fenotípica (clínica) en el 10% de los
casos.
Las características
clínicas de todas las talasemias son similares, pero su gravedad es variable. La
talasemia b minor es
clínicamente asintomática. La talasemia b mayor (anemia de
Cooley) debuta con síntomas de
anemia grave, con un espacio medular notablemente expandido y con sobrecarga
transfusional y absortiva de Fe. Los pacientes tienen ictericia, úlceras en las
piernas y colelitiasis como en la anemia de células falciformes. Es frecuente la
esplenomegalia y el bazo puede ser de gran tamaño. Si existe secuestro
esplénico, se acorta el tiempo de supervivencia de los hematíes normales
transfundidos. La hiperactividad de la médula ósea provoca engrosamiento de los
huesos craneales y de las eminencias malares. La afectación de los huesos largos
es responsable de las frecuentes fracturas patológicas. El crecimiento está
alterado y la pubertad puede retrasarse significativamente o no producirse. Los
depósitos de Fe en el músculo cardíaco pueden originar disfunción e
insuficiencia cardíaca. Otra característica es la siderosis hepática, que
conduce a deterioro funcional y cirrosis. La talasemia a1
(rasgo) tiene una presentación similar a la de la talasemia b minor. Los
pacientes con hemoglobinopatía H suelen tener anemia hemolítica
sintomática y esplenomegalia.
Cómo volví a ser una mujer feliz
ResponderEliminarCon lágrimas de alegría y felicidad estoy dando mi testimonio a todos los televidentes en línea, mi problema con el cáncer de estómago etapa IB y el VIH me ha causado muchos dolores y tristezas, especialmente en mi familia.
Tenía tanto miedo de perder mi vida, sufrí la vergüenza de visitar
terapia cientos de veces, lamentablemente no encontraron una solución definitiva a mi problema, lloré todo el día y la noche, ¿tengo que vivir mi vida de esta manera? Busqué toda la atención en Internet, fui estafado por estafadores de Internet sin números... hasta que una amiga mía que se queda en el Reino Unido me presentó a una amiga suya que se curó de la misma enfermedad, y ella me presentó al Dr. Itua quien la curó de cáncer de mama por este correo electrónico drituaherbalcenter@gmail.com. www.drituaherbalcenter.com Me comuniqué con él y me prometió que todo estaría bien y tuve fe. Me envió sus medicamentos a base de hierbas a través del servicio de mensajería y me instruyó sobre cómo beberlo durante tres semanas para curar, seguí las instrucciones dadas. a mí y hoy soy una mujer feliz de nuevo. Él cura todo tipo de enfermedades.